We utilize the largest biobank of well-characterized atherosclerotic plaques with more than 4000 included patients – the Athero-Express study. We generate various types of complex “-omics” datasets, including genotyping, transcriptomics (bulk, single-cell and spatially resolved), DNA methylation (bulk and single-cell) and proteomics. These datasets are integrated with histological and clinical data to unravel mechanisms in plaque pathogenesis and allow us to identify specific processes that lead to the severe manifestation of atherosclerotic disease like stroke and myocardial infarction. Next to computational tools, we use primary in vitro model systems derived from vascular and plaque cells and molecular biology techniques for validation and mechanistic studies.
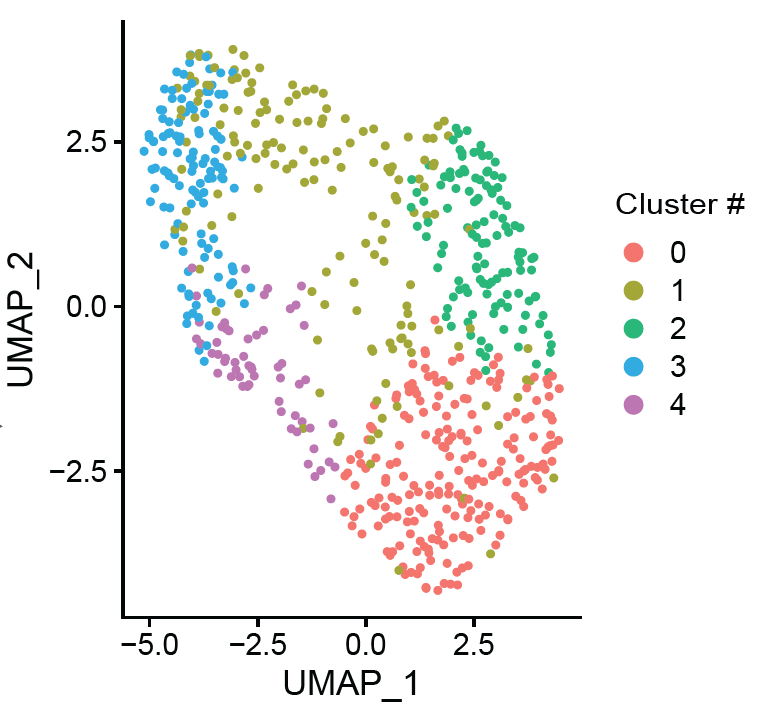
Histopathological studies have revealed key processes of atherosclerotic plaque thrombosis. However, the diversity and complexity of lesion types highlight the need for improved sub- phenotyping. We hypothesized that unbiased clustering of plaques based on gene expression results in an alternative categorization of late-stage atherosclerotic lesions.
We analyzed the gene expression profiles of 654 advanced human carotid plaques. The unsupervised, transcriptome-driven clustering revealed five dominant plaque types. These novel plaque phenotypes associated with clinical presentation (p<0.001) and showed differences in cellular compositions. Validation in coronary segments showed that the molecular signature of these plaques was linked to coronary ischemia. One of the plaque types with most severe clinical symptoms pointed to both inflammatory and fibrotic cell lineages. This highlighted plaque phenotype showed high expression of genes involved in active inflammatory processes, neutrophil degranulation, matrix turnover, and metabolism. For clinical translation, we did a first promising attempt to identify circulating biomarkers that mark these newly identified plaque phenotypes.
Preprint of this study can be found here.
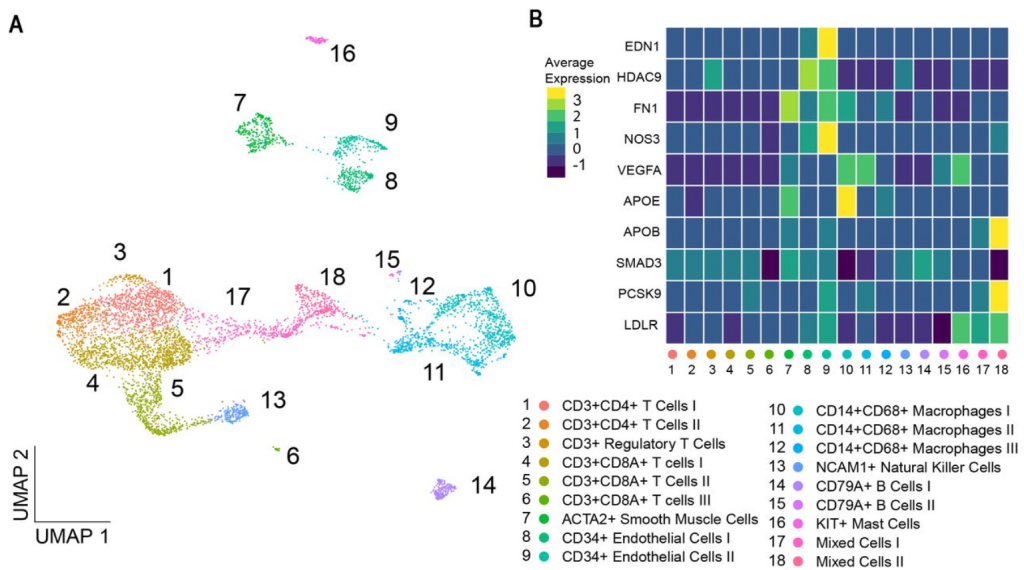
Genome-wide association studies have discovered hundreds of common genetic variants for atherosclerotic disease and cardiovascular risk factors. The translation of susceptibility loci into biological mechanisms and targets for drug discovery remains challenging. Intersecting genetic and gene expression data has led to the identification of candidate genes. However, previously studied tissues are often non-diseased and heterogeneous in cell composition, hindering accurate candidate prioritization. Therefore, we analyzed single-cell transcriptomics from atherosclerotic plaques for cell-type-specific expression to identify atherosclerosis-associated candidate gene-cell pairs.
To identify disease-associated genes, we applied gene-based analyses using GWAS summary statistics from 46 atherosclerotic and cardiovascular disease, risk factors, and other traits. We then intersected these candidates with scRNA-seq data to identify genes specific for individual cell (sub)populations in atherosclerotic plaques. The coronary artery disease loci demonstrated a prominent signal in plaque smooth muscle cells (SKI, KANK2, SORT1) p-adj. = 0.0012, and endothelial cells (SLC44A1, ATP2B1) p-adj. = 0.0011. Further sub clustering revealed genes in risk loci for coronary calcification specifically enriched in a synthetic smooth muscle cell population. Finally, we used liver-derived scRNA-seq data and showed hepatocyte-specific enrichment of genes involved in serum lipid levels.
We discovered novel gene-cell pairs, on top of known pairs, pointing to new biological mechanisms of atherosclerotic disease. We highlight that loci associated with coronary artery disease reveal prominent association levels in mainly plaque smooth muscle and endothelial cell populations. We present an intuitive single-cell transcriptomics-driven workflow rooted in human large-scale genetic studies to identify putative candidate genes and affected cells associated with cardiovascular traits. Collectively, our workflow allows for the identification of cell-specific targets relevant for atherosclerosis and can be universally applied to other complex genetic diseases and traits.
Preprint of this study can be found here.
